February 5, 2025 – by Santina Russo
Alzheimer’s disease and Parkinson’s disease are among the most prevalent neurodegenerative disorders worldwide. Recent research estimates that 12 million people currently live with Parkinson's disease. Alzheimer’s is even more common, with 7,24 million new cases recorded and more than 1,6 million deaths linked to it in 2019 alone.
Pathological features of both Alzheimer’s and Parkinson’s include the accumulation of misfolded protein aggregates in the brain. This causes a progressive loss of neuronal function, ultimately resulting in slowed information processing, memory loss, and diminished motor skills. And while both neurodegenerative disorders profoundly affect millions of individuals and their families each year, there is still no cure.
Interestingly, certain molecules are known to interact with the outer membrane of neurons and protect them from the toxic effects of the protein aggregates. Though the mechanism of this protective action remains elusive, it could be key to a breakthrough treatment. Now, with the help of CSCS’s supercomputer “Piz Daint”, scientists at the Dalle Molle Institute for Artificial Intelligence, a joint institute of the Università della Svizzera italiana (USI) and University of Applied Sciences and Arts of Southern Switzerland (SUPSI), and the University of Florence have developed a novel computational approach to investigate the membrane interaction of these and other biologically relevant molecules.
Analysing the natural protection
Specifically, the team’s application calculates the free-energy landscape of the binding process, revealing where and how molecules are most likely to insert into the membrane. The two molecules known to provide protection against toxic amyloid aggregates are the aminosterols trodusquemine (TRO) and squalamine (SQ), so both compounds are being studied intensively for their potential in Alzheimer’s and Parkinson’s treatment.
“Identifying the detailed mechanisms of any potential drug’s insertion into a membrane remains an intricate challenge,” says Gianvito Grasso, researcher at SUPSI and last author of the paper describing the new application. “In biologically relevant phenomena, such as the incorporation of a molecule into a cell membrane, your method must be able to overcome energy barriers. Without this, the events would not occur in simulations.” The scientists therefore need to add energy to their system to overcome the barrier, but where?
“Energy can be added to various variables, such as those describing the molecule’s distance to the cell membrane, its orientation, or the angles between its atoms,” says Grasso. “Typically, selecting suitable collective variables is a challenging trial-and-error process.” In the past, scientists would select one set of collective variables and run the simulation, trying to understand if the resulting molecular mechanism makes sense. In contrast, Grasso’s application uses a sampling method enhanced by machine learning (ML) named deep-TICA. Starting from hundreds or thousands of potential collective variables, the ML approach identifies the most relevant ones. In this case, Grasso and his colleagues optimized this procedure for capturing the translocation of small molecules into a cell membrane.
Essentially, the ML method determines which of the many potential collective variables best represent the slow biological process being studied. “Biological phenomena such as protein folding, protein-inhibitor binding, or the translocation of a molecule into a membrane are comparatively slow processes,” Grasso explains. “The deep-TICA method’s power lies in its ability to reduce the number of variables required to describe the system without losing accuracy in representing the slow biological dynamics.”
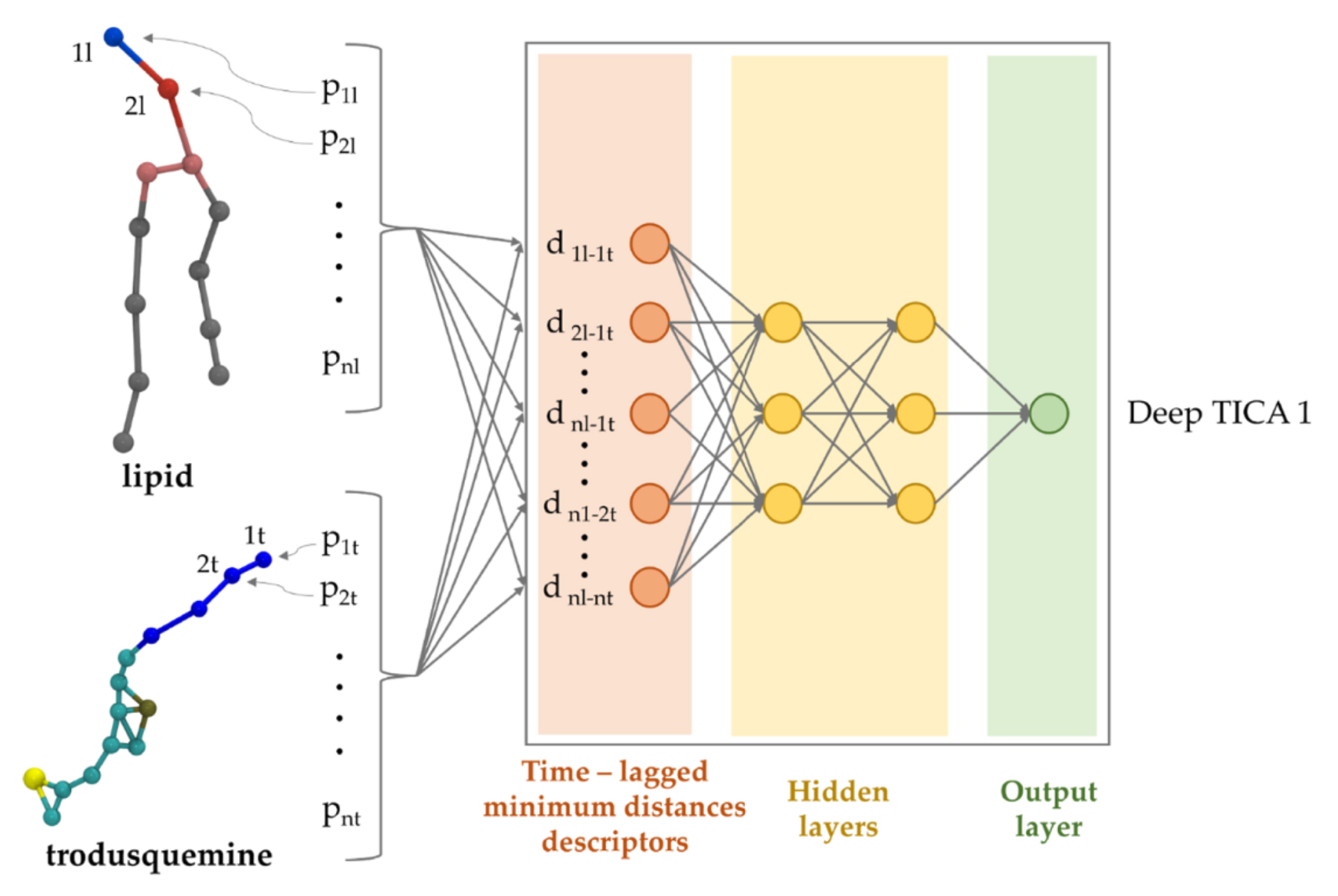
A generalized approach to membrane binding
By using the method to analyse the membrane binding of the two aminosterol molecules in simulations on the “Piz Daint” supercomputer, Grasso’s team traced the binding pathway to a neuron-specific cell membrane and calculated the binding affinities for the molecules. Squalamine demonstrated a stronger binding affinity to the neuron membrane than trodusquemine — a result confirmed by experimental data. The team is now applying their approach to screen novel molecules for their potential to offer an enhanced protection against the toxic aggregates linked to Alzheimer’s and Parkinson’s diseases.
However, the true strength of the application lies in its versatility: It can be used to analyse the membrane binding of any potential pharmaceutical drug, including peptides and proteins. Elucidating these interactions is crucial for drug design, as many diseases are linked to membrane proteins.
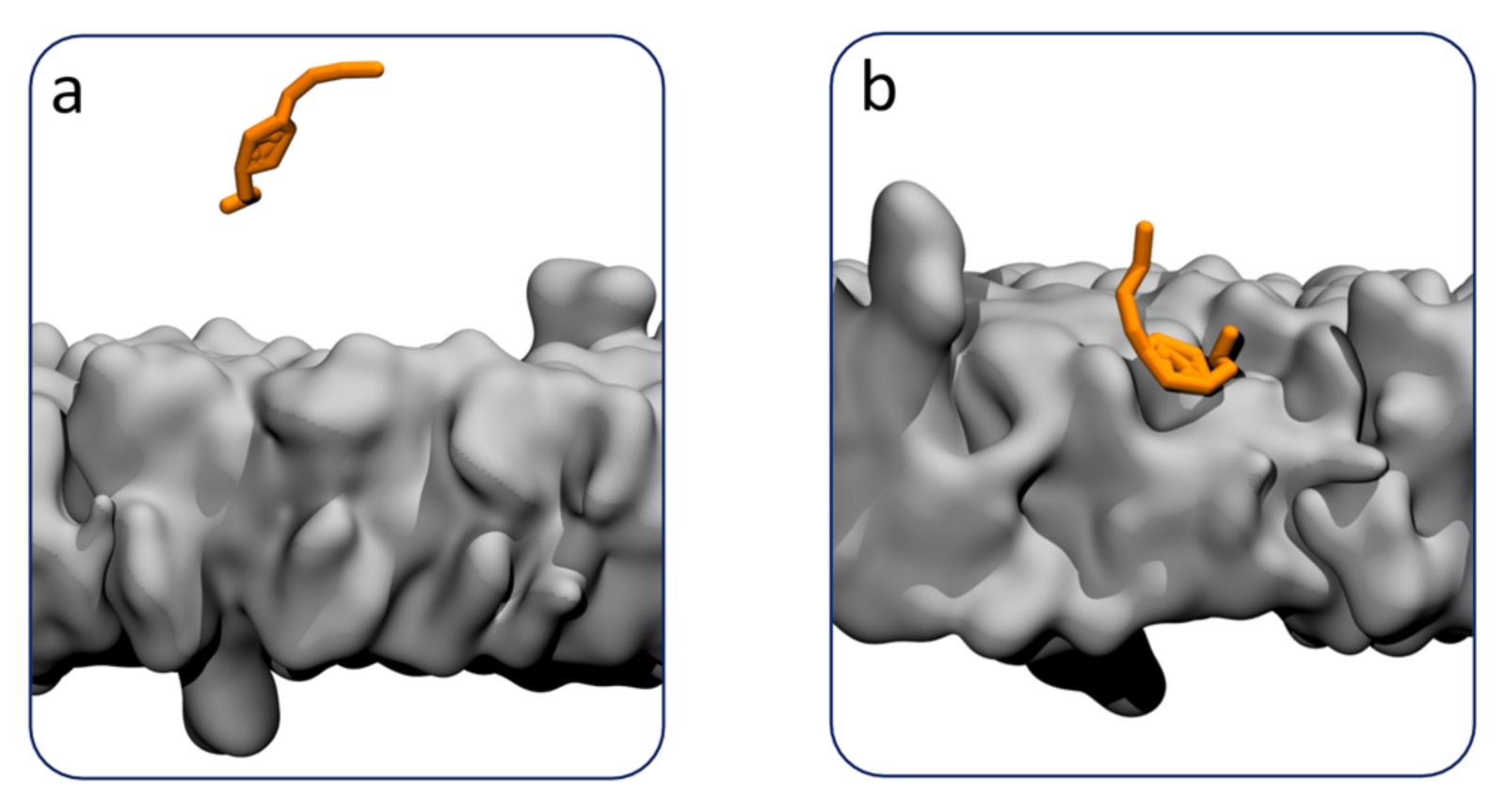
Especially influential is the family of G protein-coupled receptors (GPCRs). And as recent findings suggest, GPCRs interact with molecules that are already inserted into the membrane and reach their GPCR-binding site via lateral diffusion along the membrane. “A comprehensive understanding of how potential drug molecules bind to cell membranes is therefore key in the search for treatment for many diseases,” says Grasso. Looking ahead, the application may also help investigate interactions between molecules and the blood-brain barrier, a crucial challenge for drugs targeting the brain.
Reference:
S. Muscat, S. Errico, A. Danani, F. Chiti and G. Grasso: Leveraging Machine Learning-Guided Molecular Simulations Coupled with Experimental Data to Decipher Membrane Binding Mechanisms of Aminosterols, J. Chem. Theory Comput. (2024), 20, 18, 8279–8289, DOI: https://pubs.acs.org/doi/10.1021/acs.jctc.4c00127