September 15, 2021 - by Simone Ulmer
In countries where sufficient vaccine is available, almost all those willing have already been vaccinated against COVID-19 disease. However, there are people who cannot be vaccinated due to specific pre-existing conditions. And, in rare cases, vaccine breakthroughs happen, in which people become infected despite being vaccinated. For all of these people, it is vital to find drugs that combat the coronavirus SARS-CoV-2 and protect them against severe illness. Since the search for compounds that bind to the virus and render it harmless is laborious, calculations with supercomputers are essential to complement experiments. They facilitate the selection of possible active substances and help the pharmaceutical industry to focus on the most promising substances in drug development.
Interrupting the life cycle of the virus
Recently, a research group led by Andrew Hung, Senior Lecturer at the College of Science, Engineering and Health at RMIT University, Melbourne, and Tom Karagiannis from Monash University, Melbourne, used the CSCS supercomputer "Piz Daint" to look for such compounds: specifically, substances of low molecular weight compounds — small molecules — that can deactivate key enzymes that otherwise maintain the life cycle of the virus. Such key enzymes are the SARS-CoV-2 main protease (Mpro) and the papain-like protease (PLpro). Both of these enzymes process and ‘prepare’ certain proteins in the virus that are essential for its replication, and thus help the virus to spread.
In one of the numerous publications the researchers have published since the outbreak of the pandemic, they describe the results of computationally intensive computer simulations that enabled them to screen over 300 substances for their properties. The substances were already known and studied for their antioxidant, anti-inflammatory and antidepressant effects. Of these, 211 were phenolic compounds and 13 were fatty acids of the olive tree plant; another 76 were known pharmacological components, among them antibiotics, anti-inflammatories, and anti-malaria drugs.
First, the team looked for molecules that bind to the enzyme Mpro. The researchers made an initial preselection based on the binding affinity of the compounds in question. They filtered out all compounds that had a lower binding affinity than the one of a reference binding molecule that had already been identified by another research group. In addition, substances that are difficult to obtain were also left aside.
From olive trees to rose blooms
Based on the simulations, out of the initial 300 candidates, the scientists selected a set of 30 substances showing the strongest binding affinity to Mpro. A further narrowing down revealed that above all, hypericin, a plant constituent of St. John's wort, and cyanidin-3-O-glucoside, which occurs in hibiscus and rose plants, possess the strongest binding affinity of all compounds examined. The laboratory experiments conducted in parallel confirmed these findings. The researchers acknowledge that both hypericin and cyanidin-3-O-glucoside are not as potent as the already existing broad-spectrum antiviral drug GC376; but due to the large volume of existing research work on the two substances, they suggest that it is nevertheless worthwhile to further pursue this approach.
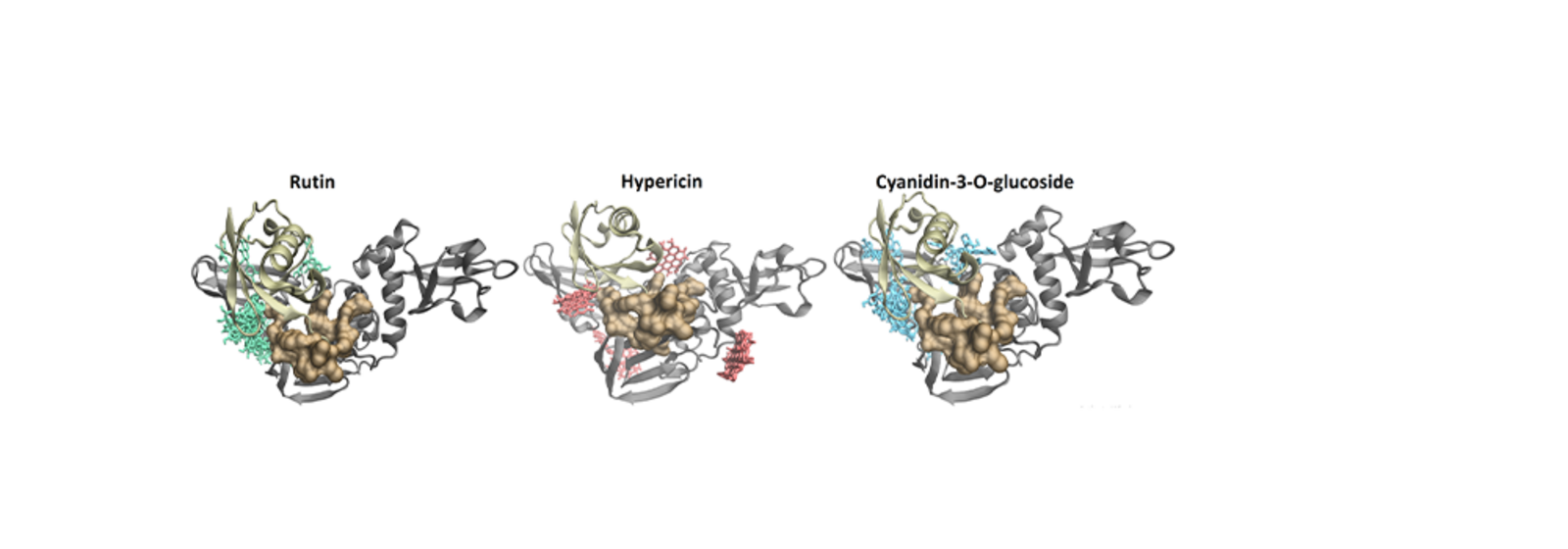
For the enzyme PLpro, the researchers found that, in addition to hypericin and cyanidin-3-O-glucoside, another compound showed potential as an antiviral substance: rutin, which occurs in plants and is often used as an antioxidant. PLpro is an attractive drug target because of its dual role. The enzyme is not only responsible for the replication of the virus, but also has a so-called deubiquitinating activity. This means that it can deactivate the small protein ubiquitin, a process which may contribute to the virus’ ability to evade host immune responses.

Different computational approaches reveal new areas of interest
The active form of the main protease Mpro is a dimer, a protein complex consisting of two almost identical subunits, with two active sites. In a further study, the researchers led by Hung and Karagiannis used a series of different computational methods at the molecular level to identify even more, previously under-studied regions of Mpro to which drugs could bind. One method is based on so-called blind docking. Instead of specifically examining a certain region for its binding ability, the method screens the entire active region of the enzyme. This way, the researchers were able to identify a new, preferred binding site for substances in the dimer — in the so-called dimer interface domain III.
In the wake of the pandemic, research groups around the world are looking for drugs to combat the viral infection. "These publications are significant because they highlight the way in which computational theoretical studies may guide lab work, potentially fast-tracking drug discovery efforts for treating COVID-19 and other diseases," concludes Hung.
Image above: The illustrations show the predicted binding poses for the dietary compounds rutin (left), hypericin (centre) and cyaniding-3-O-glucoside (right) at the PLpro enzyme (shown as grey ribbons) in the presence of ubiquitin (copper ribbons). The images highlighting the potential mechanism by which these small molecules may disrupt deubiquitinase activity. (Image: Andrew Hung)
References:
- Pitsillou E, Liang J, Karagiannis C, Ververisa K, Darmawan KK, Ng K, Hung A and Karagiannis TC: Interaction of small molecules with the SARS-CoV-2 main protease in silico and in vitro validation of potential lead compounds using an enzyme-linked immunosorbent assay, Comput. Biol. Chem.,Volume 89, December 2020, doi.org/10.1016/j.compbiolchem.2020.107408
- Pitsillou E, Liang J, Ververis K, Lim KW, Hung A and Karagiannis TC (2020): Identification of Small Molecule Inhibitors of the Deubiquitinating Activity of the SARS-CoV-2 Papain-Like Protease: in silico Molecular Docking Studies and in vitro Enzymatic Activity Assay, Front. Chem. doi.org/10.3389/fchem.2020.623971
- Lianga J, Karagiannisb C, Pitsilloua E, Darmawan KK, Ng K, Hung A, and Karagiannis TC: Site mapping and small molecule blind docking reveal a possible target site on the SARS-CoV-2 main protease dimer interface: Comput. Biol. Chem. (2020), doi.org/10.1016/j.compbiolchem.2020.107372
This article may be used on other media and online portals provided the copyright conditions are observed.